Медична генетика
- 19-09-2021, 16:31
- 625
10 Клас , Біологія і екологія 10 клас Шаламов
§ 36. Медична генетика
Медична генетика займається вивченням спадкових порушень розвитку і хвороб
Значна кількість порушень у роботі організму людини має певні спадкові передумови. А деякі порушення генетичного матеріалу проявляються у фенотипі одразу. Вивченням причин різноманітних генетичних патологій і умов їх виникнення, а також розробкою методів попередження й лікування генетичних хвороб займається медична генетика. Залежно від масштабу пошкодження спадкової інформації генетичні захворювання можуть бути спричинені точковими, хромосомними чи геномними мутаціями.
Серпуватоклітинна анемія виникає внаслідок точкової мутації і може бути корисною за умови поширення малярії
Прикладом захворювання людини, спричиненого точковою мутацією, є серпуватоклітинна анемія (див. рис. 2.2). У результаті поодинокої нуклеотидної заміни аденіну на тимін у гені гемоглобіну відбувається заміна глутамінової кислоти на валін в амінокислотному ланцюзі. Такий гемоглобін, що мутував, називають гемоглобіном S (від англ. sickle — серп). Еритроцити з гемоглобіном S набувають форми серпа, гірше переносять кисень, швидше та частіше руйнуються, особливо під час проходження крізь капіляри, що призводить до формування численних тромбів. Гомозиготи за цією мутацією часто гинуть від нестачі гемоглобіну в крові — анемії. Гетерозиготи не проявляють патологічних ознак, хоча в їхніх еритроцитах також міститься певна кількість гемоглобіну S.
Цікаво, що найчастіше мутантний алель трапляється в тропічному поясі й найбільше його поширення збігається з розповсюдженням малярійного плазмодія, що спричиняє найтяжчу — тропічну — малярію. Малярійний плазмодій — кров’яний паразит, частину свого життєвого циклу проводить в еритроциті, харчуючись його вмістом. За кілька днів дозрілий плазмодій руйнує еритроцит і потрапляє в плазму крові. У гетерозигот, що несуть алель гемоглобіну S, виникає стійкість до малярії. Це пов’язано з тим, що їхні еритроцити гинуть до того, як паразит встигає дозріти. Тому наявність алеля гемоглобіну S дає змогу виживати за умов бурхливої тропічної малярії. А в помірному кліматі, де малярія не поширена, цей алель не наділяє гетерозигот жодними перевагами й не є розповсюдженим (див. рис. 38.6).
При муковісцидозі порушується виділення йонів Хлору, а разом із ним і води з епітеліальних клітин
Іще одне поширене генетичне захворювання — муковісцидоз. Воно пов’язане з мутацією в гені трансмембранного хлорного каналу. У нормі цей білок наявний в епітеліальних тканинах і забезпечує виділення іонів Хлору з клітин. Слідом за Хлором рухаються йони Натрію, що призводить до збільшення концентрації солі навколо клітин. Потім за хлоридом натрію з клітин виходить вода. Таким чином відбувається, наприклад, виділення поту потовими залозами шкіри або слизу епітелієм бронх. Але при мутаціях, які спричинюють муковісцидоз, канал не функціонує, що призводить до підвищення в’язкості слизових секретів епітеліальних тканин. Таке порушення призводить до серйозних негативних наслідків для організму (рис. 36.1). Найбільших пошкоджень зазнають бронхи й легені, також уражаються різні залози (слинні, потові, підшлункова), тонкий кишківник. Тривалість життя людей з муковісцидозом — 25—30 років, але завдяки застосуванню лікувальної терапії може бути збільшена до 45 років. Єдиний вихід для людей з важкою легеневою формою — трансплантація легень. Також застосовують симптоматичне лікування, що зменшує прояв симптомів, але не впливає на основну причину муковісцидозу.
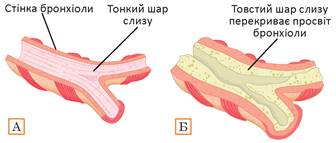
Рис. 36.1. Зміни в дихальних шляхах за муковісцидозу
А. Здорові дихальні шляхи. Б. Дихальні шляхи людини з муковісцидозом.
При синдромі Марфана зростає ризик аневризми аорти
Інше розповсюджене, але менш важке генетичне захворювання — синдром Марфана, що викликане мутацією в гені синтезу білка сполучної тканини фібрилліну.
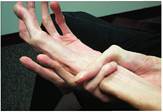
Рис. 36.2. «Павучі пальці» й надмірна гнучкість суглобів є симптомами синдрому Марфана
Воно проявляється порушенням розвитку сполучної тканини: суглоби стають дуже рухомими, а шкіра — вкрай еластичною.
Люди з синдромом Марфана худорляві, мають високий зріст, довгі руки й довгі тонкі пальці.
Небезпека цього захворювання пов’язана з тим, що може розвинутися аневризма аорти — її патологічне розширення, що супроводжується витонченням стінок. У такому стані можливий розрив аорти, що призводить до миттєвої смерті. Проте, за грамотної терапії захворювання, що призначається лікарем, пацієнт може прожити довге й щасливе життя. Багато відомих особистостей мали синдром Марфана: Авраам Лінкольн, Нікола Паганіні, Сергій Рахманінов.
Гемофілія й дальтонізм пов’язані з мутаціями в генах, розташованих у Х-хромосомі
Гемофіліями називають ряд захворювань, пов’язаних із порушенням згортання крові. У людей із гемофілією навіть невеликі порізи можуть призводити до значних втрат крові. Існує низка спадкових гемофілій. Мутації, що призводять до них, розташовуються переважно в Х-хромосомі. Нормальний алель домінує над мутантним, тому в жінок із гетерозиготним генотипом гемофілія не проявляється. Однак у чоловіків лише одна Х-хромосома і один алель цього гена в генотипі. Якщо це домінантний алель, то чоловік буде здоровий, а якщо рецесивний, то в нього буде гемофілія. Це пояснює той факт, що гемофілія набагато частіше зустрічається в чоловіків, ніж у жінок. Для того, щоб народилася жінка з гемофілією, в її батька теж повинна бути гемофілія (спробуйте пояснити, чому!), а люди з гемофілією дуже рідко доживають до репродуктивного віку. Ще одним захворюванням, пов’язаним із Х-хромосомою, є дальтонізм — порушення синтезу зорових пігментів, що призводить до порушення сприйняття кольорів. Чоловіки з дальтонізмом зустрічаються значно частіше, ніж жінки з дальтонізмом. Трохи більше про гемофілію ми поговоримо в наступному параграфі.
Синдром котячого крику зумовлений утратою ділянки п’ятої хромосоми
Не дивно, що хромосомні перебудови часто спричиняють розвиток генетичних захворювань. Утрати ділянок хромосом (делеції) часто зумовлюють загибель ембріона на ранніх стадіях розвитку, що пов’язано зі втратою численних генів, розташованих на цих ділянках. Але іноді ембріонові вдається дожити до народження. Так, делеція кінцевої ділянки п’ятої хромосоми спричиняє розвиток синдрому котячого крику. Синдром має таку дивну назву, оскільки в немовляти видозмінена будова гортані, через що плач нагадує котяче нявчання. Із віком ця ознака пропадає. У людей із цим синдромом виявляються вроджені вади серця, порушення розвитку внутрішніх органів, опорно-рухової системи, мозку. Ступінь вираженості симптомів може варіюватися: у деяких випадках відхилення несумісні з життям, в інших людина за належного лікування може дожити до 40 й навіть 50 років.
Імовірність народження дитини із синдромом Дауна залежить від віку матері
Однією з найбільш розповсюджених у людини геномних мутацій є трисомія за 21-ю хромосомою, що призводить до розвитку синдрому Дауна. Трисомія означає, що замість двох копій 21-ї хромосоми, які є в здорової людини, у людини із синдромом Дауна — три копії (рис. 36.3, А, Б). Така аномалія спричиняє розвиток патологічного стану: у дітей спостерігається зміна статури, серйозне відставання в розумовому та психічному розвитку. Проте є чимало випадків, коли люди із синдромом Дауна здобувають вищу університетську освіту, стають талановитими акторами та музикантами. Синдром Дауна — нерідкісне явище: у середньому воно трапляється один раз на 1000 вагітностей (рис. 36.3, В). Дуже важливою є залежність імовірності народження дитини із синдромом Дауна від віку матері. У жінки віком 25 років імовірність народження такої дитини близько 1:1500, віком 40 років — 1:100, віком 45 — 1:25.
Особливе місце займають генетичні захворювання, пов’язані зі зміною числа статевих хромосом
Заслуговують на увагу генетичні захворювання, що зумовлені зміною кількості статевих хромосом. Як ви пам’ятаєте, жінки мають статеві хромосоми XX, а чоловіки — XY. При моносомії за Х-хромосомою набір статевих хромосом буде Х01, і розвиватиметься синдром Шерешевського-Тернера (рис. 36.4). При цьому організм розвивається за жіночим типом — народжується дівчинка. У неї наявні численні аномалії розвитку, низький зріст, нездатність мати дітей. Щоправда, правильна гормональна терапія може значно полегшити прояв симптомів. Поєднання статевих хромосом XXY спричиняє розвиток синдрому Клайнфельтера. Організм розвивається за чоловічим типом — народжується хлопчик. Патологія зазвичай не виявляється до періоду статевого дозрівання. У цей період проявляються порушення функції статевих залоз, непропорційний розвиток молочних залоз, висока ймовірність ожиріння та розвитку цукрового діабету, безпліддя. Цікавими випадками є поєднання хромосом XYY, XYYY та XYYYY, що мають назву полісомії за Y-хромосомою. У чоловіків із таким набором хромосом немає жодних суттєвих відхилень у розвитку, але вони більш неврівноважені та схильні до агресії. Доведено, що серед ув’язнених частка чоловіків із полісомією за Y-хромосомою вища, ніж загалом у популяціях людей.
1 Є лише одна X-хромосома, відсутню хромосому в парі позначають нулем.
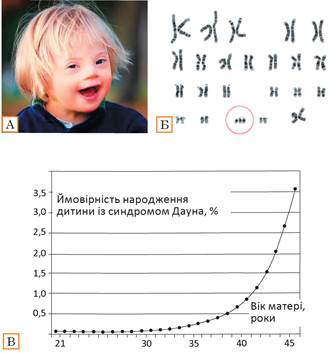
Рис. 36.3. Синдром Дауна
А. Обличчя дитини із синдромом Дауна. Б. Хромосомний набір при синдромі Дауна. В. Залежність імовірності народження дитини із синдромом Дауна від віку матері. Утім варто розуміти, що зараз у Європі, не дивлячись на відносно пізній вік народження дітей європейками, діти з синдромом Дауна майже не народжуються завдяки успіхам пренатальної діагностики.
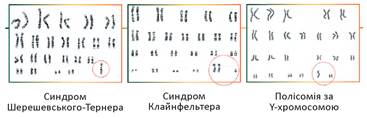
Рис. 36.4. Каріотипи людей із генетичними захворюваннями, спричиненими зміною кількості статевих хромосом
Генна терапія зможе лікувати генетичні захворювання вже найближчим часом
Як ми могли впевнитися, генетичні захворювання є вродженими порушеннями, з якими людям доводиться жити протягом усього життя. Лікування полягає в усуненні або полегшенні симптомів хвороби. У медицині такий підхід називають симптоматичним лікуванням. Він є виправданим, адже для того, щоб усунути причину захворювання, необхідно змінити генетичну інформацію у величезній кількості клітин усього організму. Але тепер, завдяки розвитку сучасних методів молекулярної й клітинної біології, таке лікування стало можливим. Сукупність підходів, спрямованих на зміну генетичної інформації в клітинах з генетичним захворюванням з метою усунення генетичного захворювання, називають генною терапією.
Значна частина генних захворювань пов’язана з тим, що в людини відсутній функціональний алель певного гена. Таку проблему можна розв’язати, штучно вводячи в людські клітини робочий ген (рис. 36.5). Зазвичай генну терапію проводять шляхом інфікування людини видозміненим вірусом: замість своєї нуклеїнової кислоти він несе ген людини. Цей ген потрапляє до ядра клітини, вбудовується в геном і починає там працювати замість дефектного гена. Таким чином, вдалося використати зброю, спрямовану проти людини — механізми вірусної атаки — для її ж зцілення. На сьогодні генну терапію вже застосовують для лікування деяких генетичних і ракових захворювань. Це є справжнім тріумфом людської думки!
При цьому не обов’язково вводити вірус безпосередньо до тіла людини. Часто достатньо виділити культуру стовбурових клітин, здійснити їх генетичну модифікацію «у пробірці», після чого повернути клітини до тіла людини. Так, у 2017 році в Бохумі (Німеччина) вперше вилікували бульозний епідермоліз — важке генетичне захворювання, що полягає в порушенні цілісності епідермісу шкіри: будь-який дотик викликає формування ран і пухирів, люди з цим захворюванням відчувають постійний біль. Спершу лікарі виділили стовбурові клітини зі шкіри хлопчика, після чого модифікували їх за допомогою використання вірусів. Потім із цих клітин було отримано пласти шкіри, що й було пересаджено хлопчикові. Через деякий час уся шкіра була відновлена. На сьогодні він здоровий і веде активний спосіб життя (рис. 36.6). Цей експеримент наразі є найбільш значним проривом у лікуванні вроджених генетичних захворювань. Будемо сподіватися, що найближчим часом з’являться нові доступні методи лікування різноманітних генетичних хвороб. А це, перш за все, залежить від успіхів розвитку біології та медицини.
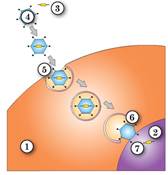
Рис. 36.5. Використання модифікованих вірусів для генної терапії спадкових захворювань
До вірусної частинки вводять фрагмент ДНК, що містить необхідний алель гена. Після інфекції клітини цей фрагмент ДНК вбудовується до геному, оскільки містить необхідні для цього елементи вірусної ДНК. Однак така вірусна частинка не може викликати захворювання, замість небезпечних генів вірусу вона несе лише потрібні для лікування захворювання гени.
1. Цитоплазма. 2. Ядро. 3. Функціональний ген. 4. Вірус-носій. 5. Ендоцитоз вірусу-носія. 6. Вивільнення вірусу-носія з везикули. 7. Потрапляння функціонального гена до ядра.
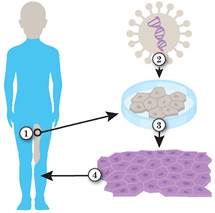
Рис. 36.6. Генна терапія бульозного епідермолізу
1. Виділення стовбурових клітин шкіри. 2. Модифікація геному стовбурових клітин шкіри лікувальним вірусом. 3. Штучне вирощування шкіри. 4. Пересадка шкіри на уражені ділянки тіла.
Окрім того, на сьогодні було розроблено й успішно введено до практики методи, що дозволяють проводити тонке редагування геному: виправлення помилок у конкретних ділянках молекули ДНК. Така система, відома як CRISPR/Cas9 (докладніше див. § 16), є молекулярною системою бактеріального противірусного імунітету. Вона дозволяє розпізнавати ділянки ДНК у живій клітині й вносити до них зміни. CRISPR/Cas9 успішно застосовується в науковій роботі дослідниками та найближчим часом буде використана й для терапії генетичних захворювань людини.
Цікаве життя
Синдром Дауна може виникнути внаслідок хромосомної мутації
Загалом синдром Дауна не э спадковим захворюванням — трисомія виникає випадково, через порушення протікання мейозу під час утворення гамет. Але іноді зустрічається сімейний синдром Дауна. Він пов’язаний із однією цікавою спадковою мутацією, що не має свого власного фенотипічного прояву. За цієї мутації довге плече 21-ї хромосоми приєднується до іншої хромосоми (частіше за все 14-ї). Людина з таким каріотипом має вигляд здорової, але в неї зростає ймовірність народження дитини з синдромом Дауна. Хоча у дитини є лише дві 21-і хромосоми, синдром все одно розвивається. Це пов’язано з наявністю додаткової порції матеріалу 21-ї хромосоми, прикріпленої до 14-ї хромосоми. Подібна мутація не мала наслідків для батьків, але призвела до народження дитини з патологією.

Життєві запитання — обійти не варто!
Елементарно про життя
• 1. Зміна форми еритроцитів при серпуватоклітинній анемії призводить до
А пришвидшення загибелі серпуватих еритроцитів
Б збільшення тривалості існування серпуватих еритроцитів
В пришвидшення розмноження малярійного плазмодія в серпуватих еритроцитах
Г сповільнення розмноження малярійного плазмодія в серпуватих еритроцитах
Д збільшення текучості крові
• 2. Слиз у бронхах і бронхіолах при муковісцидозі стає в’язким через
А збільшення концентрації йонів Хлору в слизу
Б збільшення концентрації йонів Натрію в слизу
В збільшення концентрації солі в слизу
Г недостатнє виділення води з клітин
Д надмірне виділення солі з клітин
• 3. Особа із синдромом Шерешевського-Тернера буде жіночої статі тому, що
А має в каріотипі Х-хромосому
Б не має в каріотипі Y-хромосоми
В має в каріотипі 44 аутосоми
Г люди без однієї хромосоми в парі завжди жіночої статі
Д тварини з непарною кількістю хромосом у каріотипі завжди жіночої статі
• 4. Під час генної терапії вірус
А може спричинити нове захворювання
Б може переносити функціональний алель гену
В спричиняє шкідливі мутації в геномі
Г потрапляє до ядра клітини
Д може переносити цілу хромосому
• 5. Увідповідніть порушення спадкового матеріалу й симптом, що простежується в носія цієї мутації.
1. трисомія за 21-ї хромосомою
2. мутація в гені білка, відповідального за згортання крові
3. наявність зайвої Х-хромосоми в чоловіка
4. втрата фрагменту 5-ї хромосоми
А порушення розумового та психічного розвитку
Б непропорційний розвиток молочних залоз
В порушення сприйняття кольорів
Г порушення будови гортані в немовля
Д зростання тривалості припинення кровотечі
У житті все просто
• 6. Чому існують неоднакові види гемофілій, що пов’язані з порушенням роботи різних генів?
• 7. Які порушення мейозу призводять до розвитку синдрому Дауна?
• 8. Лікування яких генетичних відхилень здається малоперспективним? Чому?
У житті все не так просто
• 9. Які небезпеки може мати генна терапія? Що потрібно для їх подолання?
• 10. Розрахуйте ймовірність народження дитини із синдромом Дауна у випадку сімейного синдрому Дауна. Мати має нормальний фенотип, а батько — транспозицію довгого плеча 21-ї хромосоми на 14-у; ембріон із моносомією по 21-й хромосомі гине задовго до народження.
Коментарі (0)